Соединения в органической химии. Названия углеводородов
Органическая химия - наука, изучающая соединения углерода с другими элементами, называемые органическими соединениями, а также законы их превращений. Название "органическая химия" возникло на ранней стадии развития науки, когда предмет изучения ограничивался соединениями углерода растительного и животного происхождения. Не все соединения углерода можно назвать органическими. Например, СО 2 , HCN, CS 2 традиционно относят к неорганическим. Условно можно считать, что прототипом органических соединений является метан СН 4 .
К настоящему времени число известных органических веществ превышает 10 млн. и увеличивается каждый год на 200-300 тыс. Многообразие этих соединений определяется уникальной способностью атомов углерода соединяться друг с другом простыми и кратными связями, образовывать соединения с практически неограниченным числом атомов, связанных в цепи, циклы, каркасы и др., образовывать прочные связи почти со всеми элементами периодичной системы, а также явлением изомерии – существованием одинаковых по составу, но разных по строению и свойствам веществ.
Огромное число органических соединений определяет значение орг. химии как крупнейшего раздела современной химии. Окружающий нас мир построен главным образом из орг. соединений; пища, топливо, одежда, лекарства, краски, моющие средства, материалы, без которых невозможно создание транспорта, книгопечатания, проникновение в космос и прочее. Важнейшую роль орг. соединения играют в процессах жизнедеятельности. По величине молекул орг. вещества делятся на низкомолекулярные (с молярной массой от нескольких десятков до нескольких сотен, редко до тысячи) и высокомолекулярные (макромолекулярные; с молярной массой порядка 10 4 -10 6 и более).
Органическая химия изучает не только соединения, получаемые из растительных и животных организмов, но в основном соединения, созданные искусственно с помощью лаборатории или промышленного органического синтеза. Более того, объектами изучения компьютерной орг. химии являются соединения, не только не существующие в живых организмах, но которые, по-видимому, нельзя получить искусственно (напр., гипотетический аналог метана, имеющий не природное тетраэдрическое строение, а форму плоского квадрата).
Историческая справка
Истоки органической химии восходят к глубокой древности (уже тогда знали о спиртовом и уксуснокислом брожении, крашении индиго и ализарином). Однако в средние века (период алхимии) были известны лишь немногие индивидуальные орг. вещества. Все исследования этого периода сводились главным образом к операциям, при помощи которых, как тогда думали, одни простые вещества можно превратить в другие. Начиная с ХVI в. (период ятрохимии) исследования были направлены в основном на выделение и использование различных лекарственных веществ: был выделен из растений ряд эфирных масел, приготовлен диэтиловый эфир, сухой перегонкой древесины получены древесный (метиловый) спирт и уксусная кислота, из винного камня – винная кислота, перегонкой свинцового сахара – уксусная кислота, перегонкой янтаря – янтарная.
Слияние химических соединений растительного и животного происхождения в единую хим. науку орг. химии осуществил Й. Берцелиус, который ввел сам термин и понятие органического вещества, образование последнего, по Берцелиусу, возможно только в живом организме при наличии "жизненной силы".
Это заблуждение опровергли Ф. Вёлер (1828), который получил мочевину (орг. вещество) из цианата аммония (неорганическое вещество), А. Кольбе, синтезировавший уксусную кислоту, М. Бертло, получивший метан из H 2 S и CS 2 , A. M. Бутлеров, синтезировавший сахаристые вещества из формалина. В первой пол. XIX в. был накоплен обширный опытный материал и сделаны первые обобщения, определившие бурное развитие орг. химии: развиты методы анализа орг. соединения (Берцелиус, Ю. Либих, Ж. Дюма, М. Шеврёль), создана теория радикалов (Вёлер, Ж. Гей-Люссак, Либих, Дюма) как групп атомов, переходящих неизменными из исходной молекулы в конечную в процессе реакции; теория типов (Ш. Жерар, 1853), в которой орг. соединения конструировались из неорганических веществ – «типов» замещением в них атомов на орг. фрагменты; введено понятие изомерии (Берцелиус).
Одновременно продолжается интенсивное развитие синтеза. Создаются первые промышленные производства органические соединения (А. Гофман, У. Перкин-старший – синтетические красители: мовеин, фуксин, цианиновые и азокрасители). Усовершенствование открытого Н. Н. Зининым (1842) способа синтеза анилина послужило основой создания анилинокрасочной промышленности.
Идея неразрывной связи хим. и физ. свойств молекулы с ее строением, идея единственности этого строения впервые была высказана Бутлеровым (1861), который создал классическую теорию хим. строения (атомы в молекулах соединяются согласно их валентностям, хим. и физ. свойства соединения определяются природой и числом входящих в их состав атомов, а также типом связей и взаимным влиянием непосредственно несвязанных атомов). Теория хим. строения определила дальнейшее бурное развитие органической химии: в 1865 Кекуле предложил формулу бензола, позднее высказал идею об осцилляции связей; В.В. Марковников и А.М. Зайцев сформулировали ряд правил, впервые связавших направление хим. реакции с хим. строением вступающего в реакцию вещества.
Работами Байера, К. Лаара, Л. Клайзена, Л. Кнорра развиты представления о таутомерии – подвижной изомерии. Все эти теоретические представления способствовали мощному развитию синтетической химии. К кон. XIX в. были получены все важнейшие представители углеводородов, спиртов, альдегидов и кетонов, карбоновых кислот, галогено- и нитропроизводных, азот- и серосодержащих структур, гетероциклов ароматической природы. Разработаны методы получения диенов, ацетиленов и алленов (А.Е. Фаворский). Открыты многочисленные реакции конденсации (Ш. Вюрц, А. П. Бородин, У. Перкин, Клайзен, А. Михаэль, Ш. Фридель, Дж. Крафтс, Э. Кнёвенагель и др.). Исключительные успехи были достигнуты Э. Г. Фишером в изучении углеводов, белков и пуринов, в использовании ферментов в орг. синтезе (1894), им же был осуществлен синтез полипептидов. Основой промышленности душистых веществ становятся работы О. Валлаха по химии терпенов. Выдающимися даже для нашего времени являются пионерские работы Р. Вильштеттера. Фундаментальный вклад в развитие орг. синтеза был внесен В. Гриньяром (1900-20) и Н.Д. Зелинским (1910) – создание исключительно плодотворного метода синтеза магнийорганических соединений и открытие каталитических превращений углеводородов; последнее сыграло выдающуюся роль в развитии химии нефти. Химия свободных радикалов началась с работ М. Гомберга (1900), открывшим трифенилметильный радикал, и была продолжена работами А. Е. Чичибабина, Г. Виланда и Ш. Гольдшмидта.
Строение органических соединений
Для органических соединений характерны неполярные ковалентные связи С-С и полярные ковалентные связи С-О, С-N, С-Hal, С-металл и т.д. Образование ковалентных связей было объяснено на основании развитых Г. Льюисом и В. Косселем (1916) предположений о важной роли электронных образований – октетов и дублетов. Молекула устойчива, если валентная оболочка таких элементов, как С, N, О, Hal, содержит 8 электронов (правило октета), а валентная оболочка водорода – 2 электрона. Хим. связь образуется обобществленной парой электронов различных атомов (простая связь). Двойные и тройные связи образуются соответствующими двумя и тремя такими парами. Электроотрицательные атомы (F, О, N) используют для связи с углеродом не все свои валентные электроны; "неиспользованные" электроны образуют неподеленные (свободные) электронные пары. Полярность и поляризуемость ковалентных связей в орг. соединениях в электронной теории Льюиса – Косселя объясняется смещением электронных пар от менее электроотрицательного к более электроотрицательному атому, что находит выражение в индуктивном эффекте и мезомерном эффекте.
Классическая теория хим. строения и первоначально электронные представления оказались не в состоянии удовлетворительно описать на языке структурных формул строение многих соединений, например, ароматических. Современная теория связи в орг. соединениях основана главным образом на понятии орбиталей и использует методы молекулярных орбиталей. Интенсивно развиваются квантовохимические методы, объективность которых определяется тем, что в их основе лежит аппарат квантовой механики, единственно пригодный для изучения явлений микромира.
Возникновение органических соединений
Большинство органических соединений в природе образуется в процессе фотосинтеза из диоксида углерода и воды под действием солнечного излучения, поглощаемого хлорофиллом в зеленых растениях. Однако орг. соединений должны были существовать на земле и до возникновения жизни, которая не могла появиться без них. Первичная земная атмосфера около 2 млрд. лет назад имела восстановительные свойства, т. к. в ней не было кислорода, а содержались прежде всего водород и вода, а также СО, азот, аммиак и метан.
В условиях сильного радиоактивного излучения земных минералов и интенсивных атмосферных разрядов в атмосфере протекал абиотический синтез аминокислот по схеме:
CH 4 + H 2 O + NH 3 →Аминокислоты
Возможность такой реакции в настоящее время доказана лабораторными опытами.
Алканы (предельные углеводороды, парафины) – ациклические насыщенные углеводороды общей формулы С n H 2n+2 . В соответствии с общей формулой алканы образуют гомологический ряд.
Первые четыре представителя имеют полусистематические названия – метан (CH 4), этан (С 2 Н 6), пропан (С 3 Н 8), бутан (С 4 Н 10). Названия последующих членов ряда строятся из корня (греческие числительные) и суффикса -ан : пентан (С 5 Н 12), гексан (С 6 Н 14), гептан (С 7 Н 16) и т. д.
Атомы углерода в алканах находятся в sp 3 -гибридном состоянии. Оси четырех sp 3 - орбиталей направлены к вершинам тетраэдра, валентные углы равны 109°28 .
Пространственное строение метана:
Энергия С-С связи Е с - с = 351 кДж/моль, длина С-С связи 0,154 нм.
Связь С-С в алканах является ковалентной неполярной. Связь С-Н – ковалентная слабополярная.
Для алканов, начиная с бутана, существуют структурные изомеры (изомеры строения), различающиеся порядком связывания между атомами углерода, с одинаковым качественным и количественным составом и молекулярной массой, но различающихся по физическим свойствам.
 Способы получения алканов
Способы получения алканов
1. С n H 2n+2 >400–700 °C > С p H 2p+2 + С m H 2m ,
Крекинг нефти (промышленный способ). Алканы также выделяют из природных источников (природный и попутный газы, нефть, каменный уголь).

(гидрирование непредельных соединений)
3. nCO + (2n + 1)Н 2 > С n H 2n+2 + nH 2 O (получение из синтез-газа (CO + Н 2))
4. (реакция Вюрца)

5. (реакция Дюма) CH 3 COONa + NaOH >t > CH 4 + Na 2 CO 3
6. (реакция Кольбе)
 Химические свойства алканов
Химические свойства алканов
Алканы не способны к реакциям присоединения, т. к. в их молекулах все связи насыщены, для них характерны реакции радикального замещения, термического разложения, окисления, изомеризации.
1. (реакционная способность убывает в ряду: F 2 > Cl 2 > Br 2 > (I 2 не идет), R 3 C > R 2 CH > RCH 2 > RCH 3 )

2. (реакция Коновалова)

3. C n H 2n+2 + SO 2 + ?O 2 >h? > C n H 2n+1 SO 3 H – алкилсульфокислота
(сульфоокисление, условия реакции: облучение УФ)
4. CH 4 >1000 °C > С + 2Н 2 ; 2CH 4 >t>1500 °C > С 2 Н 2 + ЗН 2 (разложение метана – пиролиз)
5. CH 4 + 2Н 2 O >Ni, 1300 °C > CO 2 + 4Н 2 (конверсия метана)
6. 2С n H 2n+2 + (Зn+1)O 2 > 2nCO 2 + (2n+2)Н 2 O (горение алканов)
7. 2н- С 4 Н 10 + 5O 2 > 4CH 3 COOH + 2Н 2 O (окисление алканов в промышленности; получение уксусной кислоты)
8. н- С 4 Н 10 > изо- С 4 Н 10 (изомеризация, катализатор AlCl 3)
2. Циклоалканы
Циклоалканы (циклопарафины, нафтены, цикланы, полиметилены) – предельные углеводороды с замкнутой (циклической) углеродной цепью. Общая формула С n H 2n .
Атомы углерода в циклоалканах, как и в алканах, находятся в sp 3 -гибридизованном состоянии. Гомологический ряд циклоалканов начинает простейший циклоалкан – циклопропан С 3 Н 6 , представляющий собой плоский трехчленный карбоцикл. По правилам международной номенклатуры в циклоалканах главной считается цепь углеродных атомов, образующих цикл. Название строится по названию этой замкнутой цепи с добавлением приставки «цикло» (циклопропан, циклобутан, циклопентан, циклогексан и т. д.).


Структурная изомерия циклоалканов связана с различной величиной цикла (структуры 1 и 2), строением и видом заместителей (структуры 5 и 6) и их взаимным расположением (структуры 3 и 4).
 Способы получения циклоалканов
Способы получения циклоалканов
1. Получение из дигалогенопроизводных углеводородов

2. Получение из ароматичесих углеводородов

 Химические свойства циклоалканов
Химические свойства циклоалканов
Химические свойства циклоалканов зависят от размера цикла, определяющего его устойчивость. Трех– и четырехчленные циклы (малые циклы), являясь насыщенными, резко отличаются от всех остальных предельных углеводородов. Циклопропан, циклобутан вступают в реакции присоединения. Для циклоалканов (С 5 и выше) вследствие их устойчивости характерны реакции, в которых сохраняется циклическая структура, т. е. реакции замещения.
1. Действие галогенов
2. Действие галогеноводородов
С циклоалканами, содержащими пять и более атомов углерода в цикле, галогеново-дороды не взаимодействуют.

4. Дегидрирование

Алкены (непредельные углеводороды, этиленовые углеводороды, олефины) – непредельные алифатические углеводороды, молекулы которых содержат двойную связь. Общая формула ряда алкенов С n Н 2n .
По систематической номенклатуре названия алкенов производят от названий соответствующих алканов (с тем же числом атомов углерода) путем замены суффикса – ан на – ен : этан (CH 3 -CH 3) – этен (CH 2 =CH 2) и т. д. Главная цепь выбирается таким образом, чтобы она обязательно включала в себя двойную связь. Нумерацию углеродных атомов начинают с ближнего к двойной связи конца цепи.
В молекуле алкена ненасыщенные атомы углерода находятся в sp 2 -гибридизации, а двойная связь между ними образована?– и?-связью. sp 2 -Гибридные орбитали направлены друг к другу под углом 120°, и одна негибридизованная 2р -орбиталь, расположена под углом 90° к плоскости гибридных атомных орбиталей.
Пространственное строение этилена:

Длина связи С=С 0,134 нм, энергия связи С=С Е с=с = 611 кДж/моль, энергия?-связи Е? = 260 кДж/моль.
Виды изомерии: а) изомерия цепи; б) изомерия положения двойной связи; в) Z, Е (cis, trans ) – изомерия, вид пространственной изомерии.
Способы получения алкенов1. CH 3 -CH 3 >Ni, t > CH 2 =CH 2 + H 2 (дегидрирование алканов)
2. С 2 Н 5 OH >H,SO 4 , 170 °C> CH 2 =CH 2 + Н 2 O (дегидратация спиртов)
3. (дегидрогалогенирование алкилгалогенидов по правилу Зайцева)

4. CH 2 Cl-CH 2 Cl + Zn > ZnCl 2 + CH 2 =CH 2 (дегалогенирование дигалогенопроизводных)
5. HC?CH + Н 2 >Ni, t > CH 2 =CH 2 (восстановление алкинов)
Химические свойства алкеновДля алкенов наиболее характерны реакции присоединения, они легко окисляются и полимеризуются.
1. CH 2 =CH 2 + Br 2 > CH 2 Br-CH 2 Br
(присоединение галогенов, качественная реакция)
2. (присоединение галогеноводородов по правилу Марковникова)
3. CH 2 =CH 2 + Н 2 >Ni, t > CH 3 -CH 3 (гидрирование)
4. CH 2 =CH 2 + Н 2 O >H + > CH 3 CH 2 OH (гидратация)
5. ЗCH 2 =CH 2 + 2КMnO 4 + 4Н 2 O > ЗCH 2 OH-CH 2 OH + 2MnO 2 v + 2KOH (мягкое окисление, качественная реакция)
6. CH 2 =CH-CH 2 -CH 3 + КMnO 4 >H + > CO 2 + С 2 Н 5 COOH (жесткое окисление)
7. CH 2 =CH-CH 2 -CH 3 + O 3 > Н 2 С=O + CH 3 CH 2 CH=O формальдегид+пропаналь > (озонолиз)
8. С 2 Н 4 + 3O 2 > 2CO 2 + 2Н 2 O (реакция горения)
9. (полимеризация)

10. CH 3 -CH=CH 2 + HBr >перекись > CH 3 -CH 2 -CH 2 Br (присоединение бро-моводорода против правила Марковникова)
11. (реакция замещения в?-положение)

Алкины (ацетиленовые углеводороды) – ненасыщенные углеводороды, имеющие в своем составе тройную С?С связь. Общая формула алкинов с одной тройной связью С n Н 2n-2 . Простейший представитель ряда алкинов CH?CH имеет тривиальное название ацетилен. По систематической номенклатуре названия ацетиленовых углеводородов производят от названий соответствующих алканов (с тем же числом атомов углерода) путем замены суффикса –ан на -ин : этан (CH 3 -CH 3) – этин (CH?CH) и т. д. Главная цепь выбирается таким образом, чтобы она обязательно включала в себя тройную связь. Нумерацию углеродных атомов начинают с ближнего к тройной связи конца цепи.
В образовании тройной связи участвуют атомы углерода в sp -гибридизованном состоянии. Каждый из них имеет по две sp- гибридных орбитали, направленных друг к другу под углом 180°, и две негибридных p -орбитали, расположенных под углом 90° по отношению друг к другу и к sp -гибридным орбиталям.
Пространственное строение ацетилена:

Виды изомерии: 1) изомерия положения тройной связи; 2) изомерия углеродного скелета; 3) межклассовая изомерия с алкадиенами и циклоалкенами.
Способы получения алкинов1. СаО + ЗС >t > СаС 2 + CO;
СаС 2 + 2Н 2 O > Са(OH) 2 + CH?CH (получение ацетилена)
2. 2CH 4 >t>1500 °C > HC = CH + ЗН 2 (крекинг углеводородов)
3. CH 3 -CHCl 2 + 2KOH >в спирте > HC?CH + 2KCl + Н 2 O (дегалогенирова-ние)
CH 2 Cl-CH 2 Cl + 2KOH >в спирте > HC?CH + 2KCl + Н 2 O
Химические свойства алкиновДля алкинов характерны реакции присоединения, замещения. Алкины полиме-ризуются, изомеризуются, вступают в реакции конденсации.
1. (гидрирование)
2. HC?CH + Br 2 > CHBr=CHBr;
CHBr=CHBr + Br 2 > CHBr 2 -CHBr 2 (присоединение галогенов, качественная реакция)
3. CH 3 -С?CH + HBr > CH 3 -CBr=CH 2 ;
CH 3 -CBr=CH 2 + HBr > CH 3 -CBr 2 -CHg (присоединение галогеноводородов по правилу Марковникова)
4. (гидратация алинов, реация Кучерова)


5.(присоединение спиртов)
6.(присоединение карбоновых ислот)

7. CH?CH + 2Ag 2 O >NH 3 > AgC?CAgv + H 2 O (образование ацетиленидов, качественная реакция на концевую тройную связь)
8. CH?CH + [О] >КMnO 4 > HOOC-COOH > HCOOH + CO 2 (окисление)
9. CH?CH + CH?CH > CH 2 =CH-С?CH (катализатор – CuCl и NH 4 Cl, димеризация)
10. 3HC?CH >C, 600 °C > С 6 Н 6 (бензол) (циклоолигомеризация, реакция Зелинского)
5. Диеновые углеводороды
Алкадиены (диены) – непредельные углеводороды, молекулы которых содержат две двойные связи. Общая формула алкадиенов С n Н 2n _ 2 . Свойства алкадиенов в значительной степени зависят от взаимного расположения двойных связей в их молекулах.
Способы получения диенов1. (метод СВ. Лебедева)

2. (дегидратация)

3. (дегидрирование)
 Химические свойства диенов
Химические свойства диенов
Для сопряженных диенов характерны реакции присоединения. Сопряженные диены способны присоединять не только по двойным связям (к C 1 и С 2 , С 3 и С 4), но и к концевым (С 1 и С 4) атомам углерода с образованием двойной связи между С 2 и С 3 .


6. Ароматические углеводороды
Арены, или ароматические углеводороды, – циклические соединения, молекулы которых содержат устойчивые циклические группы атомов с замкнутой системой сопряженных связей, объединяемые понятием ароматичности, которая обуславливает общие признаки в строении и химических свойствах.
Все связи С-С в бензоле равноценны, их длина равна 0,140 нм. Это означает, что в молекуле бензола между углеродными атомами нет чисто простых и двойных связей (как в формуле, предложенной в 1865 г. немецким химиком Ф. Кекуле), а все они выровнены (дел окал изованы).
 формула Кекуле
формула Кекуле
Гомологи бензола – соединения, образованные заменой одного или нескольких атомов водорода в молекуле бензола на углеводородные радикалы (R): С 6 Н 5 -R, R-С 6 Н 4 -R. Общая формула гомологического ряда бензола С n Н 2n _ 6 (n > 6). Для названия ароматических углеводородов широко используются тривиальные названия (толуол, ксилол, кумол и т. п.). Систематические названия строят из названия углеводородного радикала (приставка) и слова «бензол» (корень): С 6 Н 5 -CH 3 (метилбензол), С 6 Н 5 -С 2 Н 5 (этилбензол). Если радикалов два или более, их положение указывается номерами атомов углерода в кольце, с которыми они связаны. Для дизамещен-ных бензолов R-С 6 Н 4 -R используется также и другой способ построения названий, при котором положение заместителей указывают перед тривиальным названием соединения приставками: орто– (o -) – заместители соседних атомов углерода кольца (1,2-); мета– (м -) – заместители через один атом углерода (1,3-); пара– (п -) – заместители на противоположных сторонах кольца (1,4-).

Виды изомерии (структурная): 1) положения заместителей для ди-, три– и тетра-замещенных бензолов (например, о-, м- и п -ксилолы); 2) углеродного скелета в боковой цепи, содержащей не менее 3 атомов углерода; 3) заместителей (R), начиная с R=С 2 Н 5 .
Способы получения ароматических углеводородов1. С 6 Н 12 >Pt, 300 °C > С 6 Н 6 + ЗН 2 (дегидрирование циклоалканов)
2. н- С 6 Н 14 >Cr 2 O 3 , 300 °C > С 6 Н 6 + 4Н 2 (дегидроциклизация алканов)
3. ЗС 2 Н 2 >С, 600 °C > С 6 Н 6 (циклотримеризация ацетилена, реакция Зелинского)
Химические свойства ароматических углеводородовПо химическим свойствам арены отличаются от предельных и непредельных углеводородов. Для аренов наиболее характерны реакции, идущие с сохранением ароматической системы, а именно реакции замещения атомов водорода, связанных с циклом. Другие реакции (присоединение, окисление), в которых участвуют делокали-зованные С-С связи бензольного кольца и нарушается его ароматичность, идут с трудом.
1. C 6 H 6 + Cl 2 >AlCl 3 > C 6 H 5 Cl + HCl (галогенирование)
2. C 6 H 6 + HNO 3 >H 2 SO 4 > C 6 H 5 -NO 2 + H 2 O (нитрование)

3. С 6 Н 6 >H 2 SO 4 > С 6 Н 5 -SO 3 H + H 2 O (сульфирование)
4. С 6 Н 6 + RCl >AlCl 3 > С 6 Н 5 -R + HCl (алкилирование)
5. (ацилирование)

6. С 6 Н 6 + ЗН 2 >t, Ni > С 6 Н 12 циклогексан (присоединение водорода)
7. (1,2,3,4,5,6-гексахлороциклогексан, присоединение хлора)

8. С 6 Н 5 -CH 3 + [О] > С 6 Н 5 -COOH кипячение с раствором КMnO 4 (окисление алкилбензолов)
7. Галогеноуглеводороды
Галогеноуглеводородами называются производные углеводородов, в которых один или несколько атомов водорода заменены на атомы галогена.
Способы получения галогеноуглеводородов1. CH 2 =CH 2 + HBr > CH 3 -CH 2 Br (гидрогалогенирование ненасыщенных углеводородов)
CH?CH + HCl > CH 2 =CHCl
2. CH 3 CH 2 OH + РCl 5 > CH 3 CH 2 Cl + POCl 3 + HCl (получение из спиртов)
CH 3 CH 2 OH + HCl > CH 3 CH 2 Cl + Н 2 O (в присутствии ZnCl 2 , t°C )
3. а) CH 4 + Cl 2 >hv> CH 3 Cl + HCl (галогенирование углеводородов)
 Химические свойства галогеноуглево-дородов
Химические свойства галогеноуглево-дородов
Наибольшее значение для соединений этого класса имеют реакции замещения и отщепления.
1. CH 3 CH 2 Br + NaOH (водн. р-р) > CH 3 CH 2 OH + NaBr (образование спиртов)
2. CH 3 CH 2 Br + NaCN > CH 3 CH 2 CN + NaBr (образование нитрилов)
3. CH 3 CH 2 Br + NH 3 > + Br --HBr - CH 3 CH 2 NH 2 (образование аминов)
4. CH 3 CH 2 Br + NaNO 2 > CH 3 CH 2 NO 2 + NaBr (образование нитросоединений)
5. CH 3 Br + 2Na + CH 3 Br > CH 3 -CH 3 + 2NaBr (реакция Вюрца)
6. CH 3 Br + Mg > CH 3 MgBr (образование магнийорганических соединений, реактив Гриньяра)
7. (дегидрогалогенирование)

Спиртами называются производные углеводородов, в молекулах которых содержится одна или несколько гидроксильных групп (-OH), связанных с насыщенными атомами углерода. Группа -OH (гидроксильная, оксигруппа) является в молекуле спирта функциональной группой. Систематические названия даются по названию углеводорода с добавлением суффикса -ол и цифры, указывающей положение гидроксигруппы. Нумерация ведется от ближайшего к OH-группе конца цепи.
По числу гидроксильных групп спирты подразделяются на одноатомные (одна группа -OH), многоатомные (две и более групп -OH). Одноатомные спирты: метанол CH 3 OH, этанол С 2 Н 5 OH; двухатомный спирт: этилен-гликоль (этандиол-1,2) HO-CH 2 -CH 2 -OH; трехатомный спирт: глицерин (пропантриол-1,2,3) HO-CH 2 -CH(OH)-CH 2 -OH. В зависимости от того, с каким атомом углерода (первичным, вторичным или третичным) связана гидроксигруппа, различают спирты первичные R-CH 2 -OH, вторичные R 2 CH-OH, третичные R 3 C-OH.
По строению радикалов, связанных с атомом кислорода, спирты подразделяются на предельные, или алканолы (CH 3 CH 2 -OH), непредельные, или алкенолы (CH 2 =CH-CH 2 -OH), ароматические (С 6 Н 5 CH 2 -OH).
Виды изомерии (структурная изомерия): 1) изомерия положения OH-группы (начиная с С 3); 2) углеродного скелета (начиная с С 4); 3) межклассовая изомерия с простыми эфирами (например, этиловый спирт CH 3 CH 2 OH и диметиловый эфир CH 3 -О-CH 3). Следствием полярности связи О-Н и наличия неподеленных пар электронов на атоме кислорода является способность спиртов к образованию водородных связей.
Способы получения спиртов1. CH 2 =CH 2 + Н 2 O/Н + > CH 3 -CH 2 OH (гидратация алкенов)
2. CH 3 -CHO + Н 2 >t, Ni > С 2 Н 5 OH (восстановление альдегидов и кетонов)
3. C 2 H 5 Br + NaOH (водн.) > С 2 Н 5 OH + NaBr (гидролиз галогенопроизводных)
ClCH 2 -CH 2 Cl + 2NaOH (водн.) > HOCH 2 -CH 2 OH + 2NaCl
4. CO + 2Н 2 >ZnO, CuO, 250 °C, 7 МПа > CH 3 OH (получение метанола, промышленность)
5. С 6 Н 12 O 6 >дрожжи > 2С 2 Н 5 OH + 2CO 2 (брожение моноз)
6. 3CH 2 =CH 2 + 2KMnO 4 + 4Н 2 O > 3CH 2 OH-CH 2 OH - этиленгиликоль + 2KOH + 2MnO 2 (окисление в мягких условиях)
7. а) CH 2 =CH-CH 3 + O 2 > CH 2 =CH-CHO + Н 2 O
б) CH 2 =CH-CHO + Н 2 > CH 2 =CH-CH 2 OH
в) CH 2 =CH-CH 2 OH + Н 2 O 2 > HOCH 2 -CH(OH)-CH 2 OH (получение глицерина)
Химические свойства спиртовХимические свойства спиртов связаны с наличием в их молекулу группы -OH. Для спиртов характерны два типа реакций: разрыв связи С-О и связи О-Н.
1. 2С 2 Н 5 OH + 2Na > Н 2 + 2C 2 H 5 ONa (образование алкоголятов металлов Na, К, Mg, Al)
2. а) С 2 Н 5 OH + NaOH ? (в водном растворе не идет)
б) CH 2 OH-CH 2 OH + 2NaOH > NaOCH 2 -CH 2 ONa + 2Н 2 O
в) (качественная реакция на многоатомные спирты – образование ярко-синего раствора с гидроксидом меди)

3. а) (образование сложных эфиров)

б) С 2 Н 5 OH + H 2 SO 4 > С 2 Н 5 -О-SO 3 H + Н 2 O (на холоду)

4. а) С 2 Н 5 OH + HBr > С 2 Н 5 Br + Н 2 O
б) С 2 Н 5 OH + РCl 5 > С 2 Н 5 Cl + POCl 3 + HCl
в) С 2 Н 5 OH + SOCl 2 > С 2 Н 5 Cl + SO 2 + HCl (замещение гидроксильной группы на галоген)
5. С 2 Н 5 OH + HOC 2 H 5 >H 2 SO 4 , <140 °C > C 2 H 5 -O-C 2 H 5 + H 2 O (межмолекулярная гидротация)
6. С 2 Н 5 OH >H 2 SO 4 , 170 °C > CH 2 =CH 2 + H 2 O (внутримолекулярная гидротация)
7. а) (дегидрирование, окисление первичных спиртов)

Фенолами называются производные аренов, в которых один или несколько атомов водорода ароматического кольца замещены на гидроксильные группы. По числу гидроксильных групп в ароматическом кольце различают одно– и многоатомные (двух– и трехатомные) фенолы. Для большинства фенолов используются тривиальные названия. Структурная изомерия фенолов связана с различным положением гидроксильных групп.
 Способы получения фенолов
Способы получения фенолов
1. С 6 Н 5 Cl + NaOH(p, 340°C) > С 6 Н 5 OH + NaCl (щелочной гидролиз галогеноуглеводородов)
2. (кумольный способ получения)

3. C 6 H 5 SO 3 Na + NaOH (300–350°C) > С 6 Н 5 OH + Na 2 SO 3 (щелочное плавление солей ароматических сульфоновых кислот)
Химические свойства феноловФенолы в большинстве реакций по связи О-Н активнее спиртов, поскольку эта связь более полярна за счет смещения электронной плотности от атома кислорода в сторону бензольного кольца (участие непо-деленной электронной пары атома кислорода в системе л-сопряжения). Кислотность фенолов значительно выше, чем спиртов.
Для фенолов реакции разрыва связи С-О не характерны. Взаимное влияние атомов в молекуле фенола проявляется не только в особенностях поведения гидроксигруппы, но и в большей реакционной способности бензольного ядра.

Гидроксильная группа повышает электронную плотность в бензольном кольце, особенно в орто– и пара- положениях (+М-эффект OH-группы). Для обнаружения фенолов используется качественная реакция с хлоридом железа(III). Одноатомные фенолы дают устойчивое сине-фиолетовое окрашивание, что связано с образованием комплексных соединений железа.
1. 2С 6 Н 5 OH + 2Na > 2C 6 H 5 ONa + Н 2 (так же, как и этанол)
2. С 6 Н 5 OH + NaOH > C 6 H 5 ONa + H 2 O (в отличие от этанола)
C 6 H 5 ONa + Н 2 O + CO 2 > С 6 Н 5 OH + NaHCO 3 (фенол более слабая кислота, чем угольная)

Фенолы не образуют сложные эфиры в реакциях с кислотами. Для этого используются более реакционноспособные производные кислот (ангидриды, хлорангидриды).
4. С 6 Н 5 OH + CH 3 CH 2 OH >NaOH > С 6 Н 5 OCH 2 CH 3 + NaBr (О-алкилирование)

(взаимодействие с бромной водой, качественная реакция)
6.(нитрование разб. HNO 3 , при нитрировании конц. HNO 3 образуется 2,4,6-тринитрофенол)

7. n C 6 H 5 OH + n CH 2 O > n H 2 O + (-C 6 H 3 OH-CH 2 -) n (поликонденсация, получение фенолформальдегидных смол)
10. Альдегиды и кетоны
Альдегидами называются соединения, в которых карбонильная группа
соединена с углеводородным радикалом и атомом водорода, а кетонами – карбонильные соединения с двумя углеводородными радикалами.

Систематические названия альдегидов строят по названию соответствующего углеводорода с добавлением суффикса –аль . Нумерацию цепи начинают с карбонильного атома углерода. Тривиальные названия производят от тривиальных названий тех кислот, в которые альдегиды превращаются при окислении: Н 2 С=O – метаналь (муравьиный альдегид, формальдегид); CH 3 CH=O – этаналь (уксусный альдегид). Систематические названия кетонов несложного строения производят от названий радикалов с добавлением слова «кетон». В более общем случае название кетона строится по названию соответствующего углеводорода и суффикса –он ; нумерацию цепи начинают от конца цепи, ближайшего к карбонильной группе. Примеры: CH 3 -CO-CH 3 – диметилкетон (пропанон, ацетон). Для альдегидов и кетонов характерна структурная изомерия. Изомерия альдегидов: а) изомерия углеродного скелета, начиная с С 4 ; б) межклассовая изомерия. Изомерия кетонов: а) углеродного скелета (с С 5); б) положения карбонильной группы (с С 5); в) межклассовая изомерия.
Атомы углерода и кислорода в карбонильной группе находятся в состоянии sp 2 - гибридизации. Связь С=O сильно полярна. Электроны кратной связи С=O смещены к электроотрицательному атому кислорода, что приводит к появлению на нем частичного отрицательного заряда, а карбонильный атом углерода приобретает частичный положительный заряд.
Способы получения альдегидов и кетонов1. а) (дегидрирование, окисление первичных спиртов)
б) (дегидрирование, окисление вторичных спиртов)

2. а) CH 3 CH 2 CHCl 2 + 2NaOH >в воде > CH 3 CH 2 CHO + 2NaCl + H 2 O (гидролиз дигалогенопроизводных)
б) CH 3 СCl 2 CH 3 + 2NaOH >в воде > CH 3 COCH 3 + 2NaCl + H 2 O
3. (гидратация алкинов, реакция Кучерова)


4. (окисление этилена до этаналя)

(окисление метана до формальдегида)
CH 4 + O 2 >400–600 °C, NO > H 2 C=O + H 2 O
Химические свойства альдегидов и ке-тоновДля карбонильных соединений характерны реакции различных типов: а) присоединение по карбонильной группе; б) восстановление и окисление; в) конденсация; д) полимеризация.
1. (присоединение циановодородной кислоты, образование гидроксинитрилов)

2. (присоединение гидросулбфита натрия)

3. (восстановление)

4. (образование полуацеталец и ацеталей)


5. (взаимодействие с гидроксоламином, образование оксима ацетальдегида)

6. (образование дигалогенопроизводных)

7. (?-галогенирование в присутствии OH?)

8. (албдольная конденсация)

9. R-CH=O + Ag 2 O >NH 3 > R-COOH + 2Agv (окисление, реакция «серебряного зеркала»)
R-CH=O + 2Cu(OH) 2 > R-COOH + Cu 2 Ov, + 2H 2 O (красный осадок, окисление)
10. (окисление кетонов, жесткие условия)

11. n CH 2 =O > (-CH2-O-) n параформ n = 8-12 (полимеризация)
11. Карбоновые кислоты и их производные
Карбоновыми кислотами называются органические соединения, содержащие одну или несколько карбоксильных групп -COOH, связанных с углеводородным радикалом. По числу карбоксильных групп кислоты подразделяются на: одноосновные (монокарбоновые) CH 3 COOH (уксусная), многоосновные (дикарбоновые, трикарбоновые и т. д.). По характеру углеводородного радикала различают кислоты: предельные (например, CH 3 CH 2 CH 2 COOH); непредельные (CH 2 =CH(-COOH); ароматические (С 6 Н 5 COOH).
Систематические названия кислот даются по названию соответствующего углеводорода с добавлением суффикса –овая и слова «кислота»: HCOOH – метановая (муравьиная) кислота, CH 3 COOH – этановая (уксусная) кислота. Для карбоновых кислот характерная структурная изомерия: а) изомерия скелета в углеводородном радикале (начиная с С 4); б) межклассовая изомерия, начиная с С 2 . Возможна цис-транс-изомерия в случае непредельных карбоновых кислот. Электронная плотность?- связи в карбонильной группе смещена в сторону атома кислорода. Вследствие этого у карбонильного углерода создается недостаток электронной плотности, и он притягивает к себе неподеленные пары атома кислорода гидроксильной группы, в результате чего электронная плотность связи О-Н смещается в сторону атома кислорода, водород становится подвижным и приобретает способность отщепляться в виде протона.

В водном растворе карбоновые кислоты диссоциируют на ионы:
R-COOH - R-COО? + Н +
Растворимость в воде и высокие температуры кипения кислот обусловлены образованием межмолекулярных водородных связей.
Способы получения карбоновых кислот1. CH 3 -СCl 3 + 3NaOH > CH 3 -COOH + 3NaCl + Н 2 O (гидролиз тригалогенопроизводных)
2. R-CHO + [О] > R-COOH (окисление альдегидов и кетонов)
3. CH 3 -CH=CH 2 + CO + Н 2 O/Н + >Ni, р, t > CH 3 -CH 2 -CH 2 -COOH (оксосинтез)
4. CH 3 C?N + 2Н 2 O/ Н + > CH 3 COOH + NH 4 (гидролиз нитрилов)
5. CO + NaOH > HCOONa; 2HCOONa + H 2 SO 4 > 2HCOOH + Na 2 SO 4 (получение HCOOH)
Химические свойства карбоновых кислот и их производныхКарбоновые кислоты проявляют высокую реакционную способность и вступают в реакции с различными веществами, образуя разнообразные соединения, среди которых большое значение имеют функциональные производные: сложные эфиры, амиды, нитрилы, соли, ангидриды, гало-генангидриды.
1. а) 2CH 3 COOH + Fe > (CH 3 COO) 2 Fe + Н 2 (образование солей)
б) 2CH 3 COOH + MgO > (CH 3 COO) 2 Mg + Н 2 O
в) CH 3 COOH + KOH > CH 3 COОК + Н 2 O
г) CH 3 COOH + NaHCO 3 > CH 3 COONa + CO 2 + Н 2 O
CH 3 COONa + H 2 O - CH 3 COOH + NaOH (соли карбоновых кислот гидролизуются)
2. (образование вложных эфиров)

(омыление вложного эфира)

3. (получение хлорангидридов кислот)

4. (разложение водой)
5. CH 3 -COOH + Cl 2 >hv > Cl-CH 2 -COOH + HCl (галогенирование в?-положение)
6. HO-CH=O + Ag 2 O >NH 3 > 2Ag + Н 2 CO 3 (Н 2 O + CO 2) (особенности HCOOH)
HCOOH >t > CO + Н 2 O
Жиры – сложные эфиры глицерина и высших одноатомных карбоновых кислот. Общее название таких соединений – триглицериды. В состав природных триглицеридов входят остатки насыщенных кислот (пальмитиновой С 15 Н 31 COOH, стеариновой С 17 Н 35 COOH) и ненасыщенных (олеиновой С 17 Н 33 COOH, линолевой С 17 Н 31 COOH). Жиры состоят главным образом из триглицеридов предельных кислот. Растительные жиры – масла (подсолнечное, соевое) – жидкости. В состав триглицеридов масел входят остатки непредельных кислот.

Жирам как сложным эфирам свойственна обратимая реакция гидролиза, катализируемая минеральными кислотами. При участии щелочей гидролиз жиров происходит необратимо. Продуктами в этом случае являются мыла – соли высших карбоновых кислот и щелочных металлов. Натриевые соли – твердые мыла, калиевые – жидкие. Реакция щелочного гидролиза жиров называется также омылением.

Амины – органические производные аммиака, в молекуле которого один, два или три атома водорода замещены на углеводородные радикалы. В зависимости от числа углеводородных радикалов различают первичные RNH 2 , вторичные R 2 NH, третичные R 3 N амины. По характеру углеводородного радикала амины подразделяются на алифатические (жирные), ароматические и смешанные (или жирноароматические). Названия аминов в большинстве случаев образуют из названий углеводородных радикалов и суффикса –амин. Например, CH 3 NH 2 – метиламин; CH 3 -CH 2 -NH 2 – этиламин. Если амин содержит различные радикалы, то их перечисляют в алфавитном порядке: CH 3 -CH 2 -NH-CH 3 – ме-тилэтиламин.
Изомерия аминов определяется количеством и строением радикалов, а также положением аминогруппы. Связь N-Н является полярной, поэтому первичные и вторичные амины образуют межмолекулярные водородные связи. Третичные амины не образуют ассоциирующих водородных связей. Амины способны к образованию водородных связей с водой. Поэтому низшие амины хорошо растворимы в воде. С увеличением числа и размеров углеводородных радикалов растворимость аминов в воде уменьшается.
Способы получения аминов1. R-NO 2 + 6[Н] > R-NH 2 + 2H 2 O (восстановление нитросоединений)
2. NH 3 + CH 3 I > I? >NH 3 > CH 3 NH 2 + NH 4 I (алкилирование аммиака)
3. а) С 6 Н 5 -NO 2 + 3(NH 4) 2 S > С 6 Н 5 -NH 2 + 3S + 6NH 3 + 2H 2 O (реакция Зинина)
б) С 6 Н 5 -NO 2 + 3Fe + 6HCl > С 6 Н 5 -NH 2 + 3FeCl 2 + 2Н 2 O (восстановление нитросоединений)
в) С 6 Н 5 -NO 2 + ЗН 2 >катализатор, t > C 6 H 5 -NH 2 + 2Н 2 O
4. R-C?N + 4[H] > RCH 2 NH 2 (восстановление нитрилов)
5. ROH + NH 3 >Al 2 O 3 ,350 °C > RNH 2 + 2H 2 O (получение низших алкиламинов С 2 -С 4)
Химические свойства аминовАмины имеют сходное с аммиаком строение и проявляют подобные ему свойства. Как в аммиаке, так и в аминах атом азота имеет неподеленную пару электронов. Для аминов характерны ярко выраженные основные свойства. Водные растворы алифатических аминов проявляют щелочную реакцию. Алифатические амины – более сильные основания, чем аммиак. Ароматические амины являются более слабыми основаниями, чем аммиак, поскольку не-поделенная электронная пара атома азота смещается в сторону бензольного кольца, вступая в сопряжение с его?-электронами.

На основность аминов влияют различные факторы: электронные эффекты углеводородных радикалов, пространственное экранирование радикалами атома азота, а также способность образующихся ионов к стабилизации за счет сольватации в среде растворителя. В результате донорного эффекта алкильных групп основность алифатических аминов в газовой фазе (без растворителя) растет в ряду: первичные < вторичные < третичные. Основность ароматических аминов зависит также от характера заместителей в бензольном кольце. Электроноакцепторные заместители (-F, -Cl, -NO 2 и т. п.) уменьшают основные свойства ариламина по сравнению с анилином, а электронодонорные (алкил R-, -OCH 3 , -N(CH 3) 2 и др.), напротив, увеличивают.
1. CH 3 -NH 2 + Н 2 O > OH (взаимодействие с водой)
2. (CH 3) 2 NH + HCl > [(CH 3) 2 NH 2 ]Cl хлорид диметиламмония (взаимодействие с кислотами)
[(CH 3) 2 NH 2 ]Cl + NaOH > (CH 3) 2 NH + NaCl + H 2 O (взаимодействие солей аминов со щелочами)

(ацителирование, с третичными аминами не идет)
4. R-NH 2 + CH 3 I > I? >NH 3 > CH 3 NHR + NH 4 I (алкилирование)
5. Взаимодействие с азотистой кислотой: строение продуктов реакции с азотистой кислотой зависит от характера амина. Поэтому данная реакция используется для различия первичных, вторичных и третичных аминов.
а) R-NH 2 + HNO 2 > R-OH + N 2 + H 2 O (первичные жирные амины)
б) С 6 Н 5 -NH 2 + NaNO 2 + HCl > [С 6 Н 5 -N?N] + Cl? – соль диазония (первичные ароматические амины)
в) R 2 NH + Н-О-N=O > R 2 N-N=O (N-нитрозамин) + Н 2 O (вторичные жирные и ароматические амины)
г) R 3 N + Н-О-N=O > при низкой температуре нет реакции (третичные жирные амины)

(третичные ароматические амины)
Свойства анилина. Для анилина характерны реакции как по аминогруппе, так и по бензольному кольцу. Бензольное кольцо ослабляет основные свойства аминогруппы по сравнению с алифатическими аминами и аммиаком, но под влиянием аминогруппы бензольное кольцо становится более активным в реакциях замещения по сравнению с бензолом.
C 6 H 5 -NH 2 + HCl > Cl = C 6 H 5 NH 2 HCl
C 6 H 5 NH 2 HCl + NaOH > C 6 H 5 NH 2 + NaCl + H 2 O
C 6 H 5 NH 2 + CH3I >t > + I?

14. Аминокислоты
Аминокислотами называются гетеро-функциональные соединения, молекулы которых содержат одновременно аминогруппу и карбоксильную группу. В зависимости от взаимного расположения амино– и карбоксильной групп аминокислоты подразделяют на?-, ?-, ?– и т. д. По ИЮПАК, для наименования аминокислот группу NH 2 - называют приставкой амино-, указывая цифрой номер углеродного атома, с которым она связана, а затем следует название соответствующей кислоты.
 2-аминопропановая кислота (?-аминопропановая, ?-аланин)
2-аминопропановая кислота (?-аминопропановая, ?-аланин)
 3-аминопропановая кислота (?-аминопропановая, ?-аланин)
3-аминопропановая кислота (?-аминопропановая, ?-аланин)
 6-аминогексановая кислота (?-аминокапроновая)
6-аминогексановая кислота (?-аминокапроновая)
По характеру углеводородного радикала различают алифатические (жирные) и ароматические аминокислоты. Изомерия аминокислот зависит от строения углеродного скелета, положения аминогруппы по отношению к карбоксильной группе. Для аминокислот характерна еще оптическая изомерия.
Способы получения аминокислот1. (аммонолиз галогенокислот)

2. CH 2 =CH-COOH + NH 3 > H 2 N-CH 2 -CH 2 -COOH (присоединение аммиака к?, ?-непредельным кислотам)

(действие HCN и NH 3 на альдегиды или кетоны)
4. Гидролиз белков под влиянием ферментов, кислот или щелочей.
5. Микробиологический синтез.
Химические свойства аминокислотАминокислоты проявляют свойства оснований за счет аминогруппы и свойства кислот за счет карбоксильной группы, т. е. являются амфотерными соединениями. В кристаллическом состоянии и в среде, близкой к нейтральной, аминокислоты существуют в виде внутренней соли – дипо-лярного иона, называемого также цвиттер-ион H 3 N + -CH 2 -COO?.
1. H 2 N-CH 2 -COOH + HCl > Cl? (образование солей по аминогруппе)
2. H 2 N-CH 2 -COOH + NaOH > H 2 N-CH 2 -COO?Na + + H 2 O (образование солей)

(образование сложного эфира)

(ацилирование)
5. + NH 3 -CH 2 -COO? + 3CH 3 I >-HI > (CH 3) 3 N + -CH 2 -COO? – бетаин аминоуксусной кислоты
(алкилирование)

(взаимодействие с азотистой кислотой)
7. n H 2 N-(CH 2) 5 -COOH > (-HN-(CH 2) 5 -CO-) n + n H 2 O (получение капрона)
15. Углеводы. Моносахариды. Олигосахариды. Полисахариды
Углеводы (сахара) – органические соединения, имеющие сходное строение и свойства, состав большинства которых отражает формула С х (Н 2 O) y , где х, у ? 3.
Классификация:

Моносахариды не гидролизуются с образованием более простых углеводов. Олиго-и полисахариды расщепляются при кислом гидролизе до моносахаридов. Общеизвестные представители: глюкоза (виноградный сахар) С 6 Н 12 O 6 , сахароза (тростниковый, свекловичный сахар) С 12 Н 22 О 11 , крахмал и целлюлоза [С 6 Н 10 О 5 ] n .
Способы получения1. mCO 2 + nН 2 O >hv, хлорофилл > C m (H 2 O) n (углеводы)+ mO 2 (получение при фотосинтезе)
углеводы: С 6 Н 12 O 6 + 6O 2 > 6CO 2 + 6Н 2 O + 2920 кДж
(метаболизм: глюкоза окисляется с выделением большого количества энергии в живом организме в процессе метаболизма)
2. 6nCO 2 + 5nН 2 O >hv, хлорофилл > (С 6 Н 10 О 5) n + 6nO 2 (получение крахмала или целлюлозы)
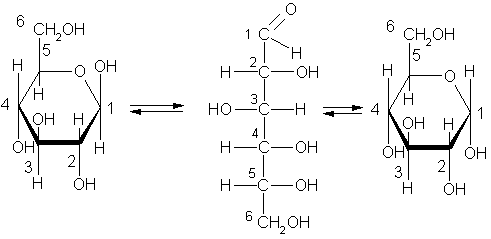
Химические свойстваМоносахриды. Все монозы в кристаллическом состоянии имеют циклическое строение (?– или?-). При растворении в воде циклический полуацеталь разрушается, превращаясь в линейную (оксо-) форму.


Химические свойства моносахаридов обусловлены наличием в молекуле функциональных групп трех видов (карбонила, спиртовых гидроксилов и гликозидного (полуацетального) гидроксила).
1. С 5 Н 11 O 5 -CHO (глюкоза) + Ag 2 O >NH 3 > CH 2 OH-(CHOH) 4 -COOH (глюконовая кислота) + 2Ag (окисление)
2. С 5 Н 11 O 5 -CHO (глюкоза) + [Н] > CH 2 OH-(CHOH) 4 -CH 2 OH(сорбит)(восстановление)

(моноалкилирование)

(полиалкилирование)

5. Важнейшим свойством моносахаридов является их ферментативное брожение, т. е. распад молекул на осколки под действием различных ферментов. Брожению подвергаются в основном гексозы в присутствии ферментов, выделяемых дрожжевыми грибками, бактериями или плесневыми грибками. В зависимости от природы действующего фермента различают реакции следующих видов:
а) С 6 Н 12 O 6 > 2С 2 Н 5 OH + 2CO 2 (спиртовое брожение);
б) С 6 Н 12 O 6 > 2CH 3 -CH(OH)-COOH (молочнокислое брожение);
в) С 6 Н 12 O 6 > С 3 Н 7 COOH + 2CO 2 + 2Н 2 O (маслянокислое брожение);
г) С 6 Н 12 O 6 + O 2 > HOOC-CH 2 -С(OH)(COOH)-CH 2 -COOH + 2Н 2 O (лимоннокислое брожение);
д) 2С 6 Н 12 O 6 > С 4 Н 9 OH + CH 3 -CO-CH 3 + 5CO 2 + 4Н 2 (ацетон-бутанольное брожение).
Дисахариды. Дисахариды – углеводы, молекулы которых состоят из двух остатков моносахаридов, соединенных друг с другом за счет взаимодействия гидроксильных групп (двух полуацетальных или одной полуацетальной и одной спиртовой). Отсутствие или наличие гликозидного (полуацетального) гидроксила отражается на свойствах дисахаридов. Биозы делятся на две группы: восстанавливающие и невосстанавливающие. Восстанавливающие биозы способны проявлять свойства восстановителей и при взаимодействии с аммиачным раствором серебра окисляться до соответствующих кислот, содержат в своей структуре гликозидный гидроксил, связь между монозами – гликозид-гликозная. Схема образования восстанавливающих биоз на примере мальтозы:

Для дисахаридов характерна реакция гидролиза, в результате которой образуются две молекулы моносахаридов:

Примером наиболее распространенных в природе дисахаридов является сахароза (свекловичный или тростниковый сахар). Молекула сахарозы состоит из остатков?-D-глюкопиранозы и?-D-фруктофуранозы, соединенных друг с другом за счет взаимодействия полуацетальных (гликозидных) гидроксилов. Биозы этого типа не проявляют восстанавливающих свойств, так как не содержат в своей структуре гликозидного гидроксила, связь между монозами – гликозид-гликозидная. Подобные дисахариды называют невосстанавливающими, т. е. не способными окисляться.
Схема образования сахарозы:

Инверсия сахарозы. При кислом гидролизе (+)сахарозы или при действии инвертазы образуются равные количества D(+)глюкозы и D(-)фруктозы. Гидролиз сопровождается изменением знака удельного угла вращения [?] с положительного на отрицательный, поэтому процесс называют инверсией, а смесь D(+)глюкозы и D(-)фруктозы – инвертным сахаром.

Полисахариды (полиозы). Полисахариды – природные высокомолекулярные углеводы, макромолекулы которых состоят из остатков моносахаридов. Основные представители: крахмал и целлюлоза, которые построены из остатков одного моносахарида – D-глюкозы. Крахмал и целлюлоза имеют одинаковую молекулярную формулу: (С 6 Н 10 О 5) n , но различные свойства. Это объясняется особенностями их пространственного строения. Крахмал состоит из остатков?-D-глюкозы, а целлюлоза – из?-D-глюкозы. Крахмал – резервный полисахарид растений, накапливается в виде зерен в клетках семян, луковиц, листьев, стеблей, представляет собой белое аморфное вещество, нерастворимое в холодной воде. Крахмал – смесь амилозы и амилопектина, которые построены из остатков?-D-глюкопиранозы.
Амилоза – линейный полисахарид, связь между остатками D-глюкозы 1?-4. Форма цепи – спиралевидная, один виток спирали содержит 6 остатков D-глюкозы. Содержание амилозы в крахмале – 15–25 %.
 амилоза
амилоза
 амилопектин
амилопектин
Амилопектин – разветвленный полисахарид, связи между остатками D-глюкозы – 1?-4 и 1?-6. Содержание амилопектина в крахмале 75–85 %.
1. Образование простых и сложных эфиров (аналогично биозам).
2. Качественная реакция – окрашивание при добавлении иода: для амилозы – в синий цвет, для амилопектина – в красный цвет.
3. Кислый гидролиз крахмала: крахмал > декстрины > мальтоза > ?-D-глюкоза.
Целлюлоза. Структурный полисахарид растений, построен из остатков?-D-глюкопиранозы, характер соединения 1?-4. Содержание целлюлозы, например, в хлопчатнике – 90–99 %, в лиственных породах – 40–50 %. Этот биополимер обладает большой механической прочностью и выполняет роль опорного материала растений, образуя стенки растительных клеток.
Характеристика химических свойств1. Кислый гидролиз (осахаривание): целлюлоза > целлобиоза > ?-D-глюкоза.
2. Образование сложных эфиров


Из растворов ацетата целлюлозы в ацетоне изготавливают ацетатное волокно.

Нитроцеллюлоза взрывоопасна, составляет основу бездымного пороха. Пироксилин – смесь ди– и тринитратов целлюлозы – используют для изготовления целлулоида, коллодия, фотопленок, лаков.
1. Реакция гидролиза или омыления. Как уже было сказано выше, реакция этерификации является обратимой, поэтому в присутствии кислот будет протекать обратная реакция, называемая гидролизом, в результате которой образуются исходные жирные кислоты и спирт: Реакция гидролиза катализируется и щелочами; в этом случае гидролиз необратим: так как получающаяся карбоновая кислота со щелочью образует соль: R – COOH…

Белки - это природные полипептиды с высокими значениями молекулярной массы (от 10 000 до десятков миллионов). Они входят в состав всех живых организмов и выполняют разнообразные биологические функции. Можно выделить четыре уровня в строении полипептидной цепи. Первичная структура белка — это конкретная последовательность аминокислот в полипептидной цепи. Пептидная цепь имеет линейную структуру только у небольшого…

Каучуки - продукты полимеризации диенов и их производных. Натуральный каучук получают из латекса - сока некоторых тропических растений. Его строение можно установить по химическим свойствам: каучук присоединяет бром, бромоводород и водород, а при нагревании без доступа воздуха распадается с образованием изопрена (2-метилбутадиена). Это означает, что каучук представляет собой непредельный полимер - полиизопрен. При более детальном…
Важнейшим из моносахаридов является глюкоза С6Н12О6, которую иначе называют виноградным сахаром. Это белое кристаллическое вещество, сладкое на вкус, хорошо растворимое в воде. Глюкоза содержится в растительных и живых организмах, в особенности велико ее содержание в виноградном соке (отсюда и название — виноградный сахар), в меде, а также в спелых фруктах и ягодах. Строение глюкозы выведено…

Физические свойства белков весьма разнообразны и определяются их строением. По физическим свойствам белки делят на два класса: глобулярные белки растворяются в воде или образуют коллоидные растворы, фибриллярные белки в воде нерастворимы. Химические свойства. 1. Разрушение вторичной и третичной структуры белка с сохранением первичной структуры называется денатурацией. Она происходит при нагревании, изменении кислотности среды, действии излучения….

Промышленный спрос на каучук значительно превосходит возможности его природных источников, поэтому химикам пришлось решать проблему синтеза каучука, не уступающего по свойствам натуральному продукту. Первый промышленный синтетический каучук был получен в России в 1931 г. Профессор С.В.Лебедев открыл экономичный способ производства бутадиена из этилового спирта и осуществил полимеризацию бутадиена по радикальному механизму в присутствии металлического натрия:…

Фруктоза — изомер глюкозы, содержится вместе с глюкозой в сладких плодах и меде. Она слаще глюкозы и сахарозы. Фруктоза является кетоноспиртом. Строение ее молекулы можно выразить формулой Имея гидроксильные группы, фруктоза, как и глюкоза, способна образовывать сахараты и сложные эфиры. Однако вследствие отсутствия альдегидной группы она в меньшей степени подвержена окислению, чем глюкоза. Фруктоза, так…
Гетероциклические соединения - органические соединения, содержащие в своих молекулах циклы, в образовании которых принимают участие неуглеродные атомы (гетероатомы). Гетероциклические соединения классифицируют по числу атомов в цикле и по типу гетероатома. В данной главе мы рассмотрим только некоторые азотсодержащие гетероциклы, производные которых имеют важное биохимическое значение. Шестичленные гетероциклы Пиридин C5H5N — простейший шестичленный ароматический гетероцикл с…
Из группы дисахаридов наибольшее значение имеет сахароза, которая иначе называется свекловичным или тростниковым сахаром. Эмпирическая формула сахарозы С12Н22О11. Велико содержание сахарозы в сахарной свекле и в стеблях сахарного тростника. Она имеется также в соке березы, клена, во многих плодах и овощах. Сахароза (обыкновенный сахар) — белое кристаллическое вещество, более сладкое, чем глюкоза, хорошо растворимое в…

Химические свойства пиридина определяются наличием ароматической системы и атома азота с неподеленной электронной парой. 1. Основные свойства. Пиридин - более слабое основание, чем алифатические амины (Кb = 1,7.10-9). Его водный раствор окрашивает лакмус в синий цвет: При взаимодействии пиридина с сильными кислотами образуются соли пиридиния: 2. Ароматические свойства. Подобно бензолу, пиридин вступает в реакции электрофильного…
Органическая химия - наука об органических соединениях и их превращениях. Термин «органическая химия» был введен шведским ученым Я. Берцелиусом в начале XIX в. До этого вещества классифицировали по источнику их получения. Поэтому в XVIII в. различали три химии: «растительную», «животную» и «минеральную». В конце XVIII в. французский химик А. Лавуазье показал, что вещества, получаемые из организмов растений и животных (отсюда их название «органические соединения»), содержат в отличие от минеральных соединений лишь немногие элементы: углерод, водород, кислород, азот, а иногда фосфор и серу. Так как углерод непременно присутствует во всех органических соединениях, органическую химию с середины XIX в. часто называют химией соединений углерода.
Способность атомов углерода образовывать длинные неразветвленные и разветвленные цепи, а также кольца и присоединять к ним другие элементы или их группы является причиной разнообразия органических соединений и того, что они по своему числу значительно превосходят неорганические соединения. Ныне известно около 7 млн. органических соединений, а неорганических - примерно 200 тыс.
После работ А. Лавуазье и до середины XIX в. химики вели интенсивный поиск новых веществ в природных продуктах и разрабатывали новые способы их превращения. Особенное внимание уделялось определению элементного состава соединений, выводу их молекулярных формул и установлению зависимости свойств соединений от их состава. Оказалось, что некоторые соединения, обладая одинаковым составом, отличаются по своим свойствам. Такие соединения назвали изомерами (см. Изомерия). Было замечено, что многие соединения в химических реакциях обмениваются остающимися без изменения группами элементов. Эти группы получили наименование радикалов, а учение, пытавшееся представить органические соединения состоящими из таких радикалов,- теории радикалов. В 40-50-х гг. XIX в. предпринимались попытки классифицировать органические соединения по типу неорганических (например, этиловый спирт С2Н5-О-Н и диэтиловый эфир С2Н5-О-С2Н5 относили к типу воды Н-О-Н). Однако все эти теории, так же как и определение элементного состава и молекулярной массы органических соединений, еще не опирались на твердый фундамент достаточно разработанного атомно-молекулярного учения. Поэтому в органической химии существовал разнобой в способах записи состава веществ, и даже такое простое соединение, как уксусная кислота, изображалось разными эмпирическими формулами: С4Н4o4, С8Н804, СгН402, из которых правильной была лишь последняя.
Только после создания русским ученым А. М. Бутлеровым теории химического строения (1861) органическая химия получила прочную научную основу, обеспечившую ее стремительное развитие в последующем. Предпосылками для ее создания послужили успехи в разработке атомно-молекулярного учения, представлений о валентности и химической связи в 50-е гг. XIX в. Эта теория позволила предсказывать существование новых соединений и их свойства. Ученые приступили к систематическому химическому синтезу предсказываемых наукой органических соединений, не встречающихся в природе. Таким образом, органическая химия стала в значительной степени химией искусственных соединений.
В первой половине XIX в. химики-органики занимались синтезом и исследованием главным образом спиртов, альдегидов, кислот и некоторых других соединений - алициклических и бензоидных (см. Алифатические соединения; Алициклические соединения). Из веществ, не встречающихся в природе, были синтезированы производные хлора, иода и брома, а также первые металлоорганические соединения (см. Элемен-тоорганические соединения). Новым источником органических соединений стала каменноугольная смола. Из нее выделены бензол, нафталин, фенол и другие бензоидные соединения, а также гетероциклические соединения - хинолин, пиридин.
Во второй половине XIX в. синтезированы углеводороды, спирты, кислоты с разветвленной углеродной цепью, начались изучение строения и синтез соединений, важных в практическом отношении (индиго, изопрен, сахара). Синтез Сахаров (см. Углеводы) и многих других соединений стал возможен после возникновения стереохимии, продолжившей развитие теории химического строения. Органическая химия первой половины XIX в. была тесно связана с фармацией - наукой о лекарственных веществах.
Во второй половине XIX в. наметился прочный союз органической химии с промышленностью, в первую очередь анилинокрасочной. Перед химиками были поставлены задачи расшифровки строения известных природных красителей (ализарин, индиго и др.), создания новых красителей, а также разработки технически приемлемых способов их синтеза. Так, в 70- 80-х гг. XIX в. возникла прикладная органическая химия.
Конец XIX - начало XX в. ознаменовались созданием новых направлений в развитии органической химии. В промышленном масштабе стал использоваться богатейший источник органических соединений - нефть, и с этим было связано бурное развитие химии алициклических соединений и вообще химии углеводородов (см. Нефтехимия). Появились практически важные каталитические методы превращения органических соединений, созданные П. Сабатьё во Франции, В. Н. Ипатьевым и позднее Н. Д. Зелинским в России (см. Катализ). Теория химического строения значительно углубилась в результате открытия электрона и создания электронных представлений о строении атомов и молекул. Были открыты и разработаны мощные методы физико-химических и физических исследований молекул, в первую очередь рентгеноструктурный анализ. Это позволило выяснить строение, а следовательно, понять свойства и облегчить синтез огромного числа орган! ческих соединений.
С начала 30-х гг. XX в. в связи с возникновением квантовой механики появились вычислительные методы, позволяющие расчетным путем делать заключения о строении и свойствах органических соединений (см. Квантовая химия).
Среди новых направлений химической науки - химия органических производных фтора, получивших большое практическое значение. В 50-х гг. XX в. возникла химия ценовых соединений (ферроцен и др.), представляющая соединительное звено между органической и неорганической химией. В практику химиков-органиков прочно вошло применение изотопов. Еще в начале XX в. были открыты свободно существующие органические радикалы (см. Радикалы свободные), а впоследствии создана химия неполнова-лентных органических соединений - ионов карбония, карбанионов, радикал-ионов, молекулярных ионов (см. Ионы). В 60-х гг. были синтезированы совершенно новые типы органических соединений, например катенаны, в которых отдельные циклические молекулы связаны друг с другом, подобно тому как изображают пять переплетенных олимпийских колец.
Органическая химия в XX в. приобрела огромное практическое значение, особенно для переработки нефти, синтеза полимеров, синтеза и изучения физиологически активных веществ. В результате из органической химии выделились в самостоятельные дисциплины такие ее направления, как нефтехимия, химия полимеров, биоорганическая химия.
Современная органическая химия имеет сложную структуру. Сердцевину ее составляет препаративная органическая химия, занимающаяся выделением из природных продуктов и искусственным приготовлением индивидуальных органических соединений, а также созданием новых методов их получения. Решить эти задачи невозможно без опоры на аналитическую химию, позволяющую судить о степени очистки, гомогенности (однородности) и индивидуальности органических соединений, предоставляющую данные об их составе и строении в изолированном состоянии, а также тогда, когда они выступают в качестве исходных веществ, промежуточных и конечных продуктов реакции. Для этой цели аналитическая химия использует различные химические, физико-химические и физические методы исследования. Сознательный подход к решению задач, стоящих перед препаративной и аналитической органической химией, обеспечивается опорой их на теоретическую органическую химию. Предметом этой науки является дальнейшая разработка теории строения, а также формулирование зависимостей между составом и строением органических соединений и их свойствами, между условиями протекания органических реакций и их скоростью и достижением химического равновесия. Объектами теоретической органической химии могут быть как нереагирующие соединения, так и соединения во время их превращений, а также промежуточные, нестойкие образования, возникающие в ходе реакций.
Такая структура органической химии сложилась под влиянием различных факторов, важнейшим из которых были и остаются запросы практики. Именно этим объясняется то, например, обстоятельство, что в современной органической химии ускоренно развивается химия гетероциклических соединений, тесно связанная с таким прикладным направлением, как химия синтетических и природных лекарственных средств.
Органическая химия - раздел химии, изучающий соединения углерода, их структуру, свойства, методы синтеза, а также законы их превращений. Органическими называют соединения углерода с другими элементами (в основном с H, N, O, S, P, Si, Ge и др.).
Уникальная способность атомов углерода связываться друг с другом, образуя цепочки различной длины, циклические структуры разного размера, каркасные соединения, соединения со многими элементами, различные по составу и строению, обусловливает многообразие органических соединений. К настоящему времени число известных органических соединений на много превышает 10 млн. и увеличивается каждый год на 250-300 тыс. Окружающий нас мир построен в основном из органических соединений, к ним относятся: пища, одежда, топливо, красители, лекарства, моющие средства, материалы для самых различных отраслей техники и народного хозяйства. Органические соединения играют ключевую роль в существовании живых организмов.
На стыке органической химии с неорганической химией, биохимией и медициной возникли химия метало- и элементорганических соединений, биоорганическая и медицинская химия, химия высокомолекулярных соеди-нений.
Основным методом органической химии является синтез. Органическая химия изучает не только соединения, полученные из растительных и животных источников (природные вещества), но в основном соединения, созданные искусственно с помощью лабораторного и промышленного синтеза.
История развития органической химии
Способы получения различных органических веществ были известны ещё с древности. Так, египтяне и римляне использовали красители растительного проис-хож-де-ния - индиго и ализарин. Многие народы владели секретами производства спиртных на-пит-ков и уксуса из сахар- и крахмалсодержащего сырья.
Во времена средневековья к этим знаниям практически ничего не прибавилось, некоторый прогресс начался только в 16-17 веках (период ятрохимии), когда путем перегонки растительных продуктов были выделены новые органические соединения. В 1769-1785 г. К.В. Шееле выделил несколько органических кислот: яблочную, винную, лимонную, галловую, молочную и щавелевую. В 1773 г. Г.Ф. Руэль выделил мочевину из человеческой мочи. Выделенные из животного и растительного сырья вещества имели между собой много общего, но отличались от неорганических соединений. Так возник термин «Органическая химия» - раздел химии, изучающий вещества, выделенные из организмов (определение Й.Я . Берцелиуса , 1807 г.). При этом полагали, что эти вещества могут быть получены только в живых организмах благодаря «жизненной силе».
Принято считать, что органическая химия как наука появилась в 1828 г., когда Ф. Вёлер впервые получил органическое вещество - мочевину - в результате упаривания водного раствора неорганического вещества - цианата аммония (NH 4 OCN). Дальнейшие экспериментальные работы продемонстрировали неоспоримые аргументы несосто-ятельности теории «жизненной силы». Так, например, А. Кольбе синтезировал уксусную кислоту, М. Бертло получил метан из H 2 S и CS 2 , а А.М. Бутлеров синтезировал сахарис-тые вещества из формалина.
В середине 19 в. продолжается бурное развитие синтетической органической хи-мии, создаются первые промышленные производства органических веществ (А. Гофман, У. Перкин-старший - синтетические красители, фуксин, цианиновые и азакрасители). Усовершенствование открытого Н.Н. Зининым (1842 г.) способа синтеза анилина послужило основой для создания анилинокрасочной промышленности. В лаборатории А. Байера были синтезированы природные красители - индиго, ализарин, индигоидные, ксантеновые и антрахиноновые.
Важным этапом в развитии теоретической органической химии стала разработка Ф.А. Кекуле теории валент-ности в 1857 г., а также классической теории химического строения А.М . Бутлеровым в 1861 г., согласно которой атомы в молекулах соединяются в соответствии с их валентностью, химические и физические свойства соединений определяются природой и числом входящих в них атомов, а также типом связей и взаимным влиянием непосредственно несвязанных атомов. В 1865 г. Ф . Кекуле предложил структурную форму-лу бензола, что стало одним из важнейших открытий в органической химии. В.В. Марковников и А.М. Зайцев сформулировали ряд правил, впервые связавших направление органических реакций со строением вступающих в них веществ. В 1875 г. Вант-Гофф и Ле Бель предложили тетраэдрическую модель атома углерода, по которой валентности углерода направлены к вершинам тетраэдра, в центре которого размещён атом углерода. На основе этой модели, в сочетании с экспериментальными исследованиями И. Вислиценуса (!873 г.), показавшего идентичность структурных формул (+)-молочной кислоты (из кислого молока) и (±)-молочной кислоты, возникла стереохимия - наука о трёхмерной ориентации атомов в молекулах, которая предсказывала в случае наличия 4 различных заместителей при атоме углерода (хиральные структуры) возможность существования пространственно-зеркальных изомеров (антиподов или энантиомеров).
В 1917 г. Льюис предложил рассматривать химическую связь с помощью электронных пар.
В 1931 г. Хюккель применил квантовую теорию для объяснения свойств небензоидных ароматических систем, чем основал новое направление в органической химии - квантовую химию. Это послужило толчком для дальнейшего интенсивного развития квантовохимических методов, в частности метода молекулярных орбиталей. Этап проникновения орбитальных представлений в органическую химию открыла теория резонанса Л. Полинга (1931-1933 г.г.) и далее работы К. Фукуи, Р. Вудворда и Р. Хофмана о роли граничных орбиталей в определении направления химических реакций.
Середина 20 в. характеризуется особенно бурным развитием органического синтеза. Это определялось открытием основополагающих процессов, таких как получе-ние олефинов с использованием илидов (Г. Виттиг , 1954 г.), диеновый синтез (О. Дильс и К. Альдер , 1928 г.), гидроборирование непредельных соединений (Г. Браун , 1959 г.), синтез нуклеотидов и синтез гена (А. Тодд , Х. Корана ). Успехи в химии метало-органических соединений во многом обязаны работам А.Н. Несмеянова и Г.А. Разуваева . В 1951 г. был осуществлен синтез ферроцена, установление «сэндвичевой» структуры которого Р. Вудвордом и Дж. Уилкинсоном положило начало химии металлоценовых соединений и вообще органической химии переходных металлов.
В 20-30 г.г. А.Е. Арбузов создает основы химии фосфорорганических соединений, что впоследствии привело к открытию новых типов физиологически активных соединений, Комплексонов и др.
В 60-80 г.г. Ч. Педерсен , Д. Крам и Ж.М. Лен разрабатывают химию краун-эфиров, криптандов и других родственных структур, способных образовывать прочные молеку-ляр-ные комплексы, и тем самым подходят к важнейшей проблеме «молекулярного узнава-ния».
Современная органическая химия продолжает своё бурное развитие. В практику органического синтеза вводятся новые реагенты, принципиально новые синтетические методы и приемы, новые катализаторы, синтезируются неизвестные ранее органические структуры. Постоянно ведется поиск органических новых биологически активных соединений. Еще многие проблемы органической химии ждут своего решения, например, детальное установление взаимосвязи структура - свойства (в том числе, биологическая активность), установление строения и стереонаправленный синтез сложных природных соединений, разработка новых регио- и стереоселективных синтетических методов, поиск новых универсальных реагентов и катализаторов.
Интерес мирового сообщества к развитию органической химии ярко проде-мон-стрирован вручением Нобелевской премии по химии 2010 г. Р. Хеку, А. Судзуки и Э. Нэгиси за работы по применению палладиевых катализаторов в органическом синтезе для формирования связей углерод - углерод.
Классификация органических соединений
В основе классификации лежит структура органических соединений. Основа описания структуры - структурная формула.
Основные классы органических соединений
Углеводороды - соединения, состоящие только из углерода и водорода. Они в свою очередь делятся на:
Насыщенные - содержат только одинарные (σ-связи) и не содержат кратные связи;
Ненасыщенные - имеют в своём составе хотя бы одну двойную (π-связь) и/или тройную связь;
С открытой цепью (алициклические);
С замкнутой цепью (циклические) - содержат цикл
К ним относятся алканы, алкены, алкины, диены, циклоалканы, арены
Соединения с гетероатомами в функциональных группах - соединения, в которых углеродный радикал R связан с функциональной группой. Такие соединения классифицируют по характеру функциональной группы:
Спирт, фенолы (содержат гидроксильную группу ОН)
Простые эфиры (содержат группировку R-O-R или R-O-R
Карбонильные соединения (сожержат группировку RR"C=O), к ним относятся альдегиды, кетоны, хиноны.
Соединения, содержащие карбоксильную группу (СООН или СООR), к ним относятся карбоновые кислоты, сложные эфиры
Элемент- и металлорганические соединения
Гетероциклические соединения - содержат гетероатомы в составе цикла. Различаются по характеру цикла (насыщенный, ароматический), по числу атомов в цикле (трех-, четырёх-, пяти-, шестичленные циклы и т.д.), по природе гетероатома, по количеству гетероатомов в цикле. Это определяет огромное разнообразие известных и ежегодно синтезируемых соединений этого класса. Химия гетероциклов представляет собой одну из наиболее увлекательных и важных областей органической химии. Достаточно сказать, что более 60% лекарственных препаратов синтетического и природного происхождения относятся к различным классам гетероциклических соединений.
Природные соединения - соединения, как правило, достаточно сложного строения, зачастую принадлежащие сразу к нескольким классам органических соединений. Среди них можно выделить: аминокислоты, белки , углеводы , алкалоиды , терпены и др.
Полимеры - вещества с очень большой молекулярной массой, состоящие из периодически повторяющихся фрагментов - мономеров.
Строение органических соединений
Органические молекулы в основном образованы ковалентными неполярными связями С-С, или ковалентными полярными связями типа С-О, C-N, C-Hal. Полярность объясняется смещением электронной плотности в сторону более электроотрицательного атома. Для описания строения органических соединений химики используют язык структурных формул молекул, в которых связи между отдельными атомами обозначаются с помощью одного (простая, или одинарная связь), двух (двойная) или трёх (тройная) валентных штрихов. Понятие валентного штриха, которое не потеряло своего значения и по сей день, ввел в органическую химию А. Купер в 1858 г
Очень существенным для понимания строения органических соединений является понятие о гибридизации атомов углерода. Атом углерода в основном состоянии имеет электронную конфигурацию 1s 2 2s 2 2p 2 , на основе которой невозможно объяснить присущую углероду в его соединениях валентность 4 и существование 4 идентичных связей в алканах, направленных к вершинам тетраэдра. В рамках метода валентных связей это противоречие разрешается введением понятия о гибридизации. При возбуждении осуществляется s →p переход электрона и последующая, так называемая, sp- гибридизация, причем энергия гибридизованных орбиталей является промежуточной между энергиями s - и p -орбиталей. При образовании связей в алканах три р -электрона взаимодействуют с одним s -электроном (sp 3 -гибридизация) и возникают 4 одинаковые орбитали, расположенные под тетраэдрическими углами (109 о 28") друг к другу. Атомы углерода в алкенах находятся в sp 2 -гибридном состоянии: у каждого атома углерода имеют три одинаковые орбитали, лежащие в одной плоскости под углом 120 о друг к другу (sp 2 -орбитали), а четвертая (р -орбиталь) перпендикулярна этой плоскости. Перекрывание р -орбиталей двух атомов углерода образует двойную (π) связь. Атомы углерода, несущие тройную связь находятся в sp -гибридном состоянии.
Особенности органических реакций
В неорганических реакциях обычно участвуют ионы, такие реакции проходят быстро и до конца при комнатной температуре. В органических реакциях часто происходят разрывы ковалентных связей с образованием новых. Как правило, эти процессы требуют особых условий: определённой температуры, времени реакции, определенных растворителей, и часто наличия катализатора. Обычно протекает не одна, а сразу несколько реакций, Поэтому при изо-бра-жении органических реакций используют не уравнения, а схемы без расчёта сте-хио-метрии. Выходы целевых веществ в органических реакциях зачастую не превышают 50%, а выделение их из реакционной смеси и очистка требуют специфических методов и приёмов. Для очистки твердых веществ, как правило, используют перекристаллизацию из специально подобранных растворителей. Жидкие вещества очищают перегонкой при атмосферном давлении или в вакууме (в зависимости от температуры кипения). Для контролем за ходом реакций, разделения сложных реакционных смесей прибегают к различным видам хроматографии [тонкослойная хроматография (ТСХ), препаративная высокоэффективная жидкостная хроматография (ВЭЖХ) и др.].
Реакции могут протекать очень сложно и в несколько стадий. В качестве промежуточных соединений могут возникать радикалы R·, карбкатионы R + , карбанионы R - , карбены:СХ 2 , катион-радикалы, анион-радикалы и другие активные и нестабильные частицы, обычно живущие доли секунды. Подробное описание всех превращений, происходящих на молекулярном уровне во время реакции, называется механизмом реакции . По характеру разрыва и образования связей различают радикальные (гомолитические) и ионные (гетеролитические) про-цессы. По типам превращений различают цепные радикальные реакции, реакции нуклеофильного (алифатического и ароматического) замещения, реакции элими-ни-ро-вания, электрофильного присоединения, электрофильного замещения, конденсации, циклизации, процессы перегруппировок и др. Реакции классифицируют также по способам их инициирования (возбуждения), их кинетическому порядку (моно-молекулярные, бимолекулярные и др.).
Определение структуры органических соединений
За всё время существования органической химии как науки важнейшей задачей было определить структуру органических соединений. Это значит узнать, какие атомы входят в состав структуры, в каком порядке и каким образом эти атомы связаны между собой и как расположены в пространстве.
Существует несколько методов решения этих задач.
- Элементный анализ заключается в том, что вещество разлагают на более простые молекулы, по количеству которых можно определить количество атомов, входящих в состав соединения. Этот метод не дает возможности установить порядок связей между атомами. Часто используется лишь для подтверждения предложенной структуры.
- Инфракрасная спектроскопия (ИК спектроскопия) и спектроскопия комбинационного рассеяния (спектроскопия КР). Метод основан на том, что вещество взаимодействует с электромагнитным излучением (светом) инфра-крас-ного диапазона (в ИК спектроскопии наблюдают поглощение, в КР спектроскопии - рассеяние излучения). Этот свет при поглощении возбуждает коле-бательные и вращательные уровни молекул. Опорными данными служат число, частота и интен-сивность колебаний молекулы, связанных с изменением дипольного момента (ИК) или поляризуемости (КР). Метод позволяет установить наличие функ-циональных групп, а также часто используется для подтверждения иден-тичности вещества с некоторым уже известным веществом путём сравнения их спектров.
- Масс-спектрометрия . Вещество при определённых условиях (электронный удар, химическая ионизация и др.) превращается в ионы без потери атомов (моле-кулярные ионы) и с потерей (осколочные, фрагментарные ионы). Метод позволяет оп-ре-делить молекулярную массу вещества, его изотопный состав, иногда наличие функциональных групп. Характер фрагментации позволяет сделать некоторые вы-во-ды об особенностях строения и воссоздать структуру исследуемого соеди-нения.
- Метод ядерного магнитного резонанса (ЯМР) основан на взаимодействии ядер, обладающих собственным магнитным моментом (спином) и помещенных во внешнее постоянное магнитное поле (переориентация спина), с переменным электромагнитным излучением радиочастотного диапазона. ЯМР представляет собой один из самых главных и информативных методов определения химической структуры. Метод используют также для изучения пространственного строения и динамики молекул. В зависимости от ядер, взаимодействующих с излучением различают, например, метод протонного резонанса ПМР, ЯМР 1 Н), позволяющий определять положение атомов водорода в молекуле. Метод ЯМР 19 F позволяет определять наличие и положение атомов фтора. Метод ЯМР 31 Р дает информацию о наличии, валентном состоянии и положении атомов фосфора в молекуле. Метод ЯМР 13 С позволяет определять число и типы углеродных атомов, он используется для изучения углеродного скелета молекулы. В отличие от первых трёх в последнем методе используется неосновной изотоп элемента, поскольку ядро основного изотопа 12 С имеет нулевой спин и не может наблюдаться методом ЯМР.
- Метод ультрафиолетовой спектроскопии (УФ спектроскопия) или спектроскопия электронных переходов. Метод основан на поглощении электро-магнитного излучения ультрафиолетовой и видимой области спектра при переходе электронов в молекуле с верхних заполненных энергетических уровней на вакант-ные (возбуждение молекулы). Чаще всего используется для определения наличия и характеристики сопряженных π-систем.
- Методы аналитической химии позволяют определять наличие некоторых функциональных групп по специфическим химическим (качественным) реакциям, факт протекания которых можно фиксировать визуально (например, появление или изменение окраски) или с помощью других методов. Помимо химических методов анализа в органической химии все большее применение находят инструментальные аналитические методы, такие как хроматография (тонкослойная, газовая, жид-костная). Почетное место среди них занимает хроматомасс-спектромерия, позво-ляющая не только оценить степень чистоты полученных соединений, но и полу-чить масс-спектральную информацию о компонентах сложных смесей.
- Методы исследования стереохимии органических соединений . С начала 80 г.г. стала очевидной целесообразность разработки нового направления в фармакологии и фармации, связанного с созданием энантиомерно чистых лекарственных средств с оптимальным соотношением терапевтической эффективности и безопасности. В настоящее время примерно 15% всех синтезируемых фармпрепаратов представ-лены чистыми энантиомерами. Отражением данной тенденции стало появление в научной литературе последних лет термина chiral switch , что в русском переводе означает ”переключение на хиральные молекулы”. В связи с этим особое значение в органической химии приобретают методы установления абсолютной конфи-гурации хиральных органических молекул и определения их оптической чистоты. Основным методом определения абсолютной конфигурации следует считать рентгеноструктурный анализ (РСА), а оптической чистоты - хроматографию на колонках с неподвижной хиральной фазой и метод ЯМР с использованием специальных дополнительных хиральных реагентов.
Связь органической химии с химической промышленностью
Основной метод органической химии - синтез - тесно связывает органическую химию с химической промышленностью. На основе методов и разработок синтетической органической химии возник малотоннажный (тонкий) органический синтез, включающий производство лекарств, витаминов, ферментов , феромонов, жидких кристаллов, орга-нических полупроводников, солнечных батарей и др. Развитие крупнотоннажного (основ-ного) органического синтеза также базируется на достижениях органической химии. К основному органическому синтезу относится производство искусственных волокон, пластмасс, переработка нефти, газа и каменноугольного сырья.
Рекомендуемая литература
- Г.В. Быков, История органической химии , М.: Мир, 1976 (http://gen.lib/rus.ec/get?md5=29a9a3f2bdc78b44ad0bad2d9ab87b87)
- Дж. Марч, Органическая химия: реакции, механизмы и структура , в 4 томах, М.: Мир, 1987
- Ф. Кери, Р. Сандберг, Углубленный курс органической химии , в 2 томах, М.: Химия, 1981
- О.А. Реутов, А.Л. Курц, К.П. Бутин, Органическая химия , в 4 частях, М.: « Бином, Лаборатория знаний», 1999-2004. (http://edu.prometey.org./library/autor/7883.html)
- Химическая энциклопедия , под ред. Кнунянца, М.: «Большая Российская энциклопедия», 1992.



